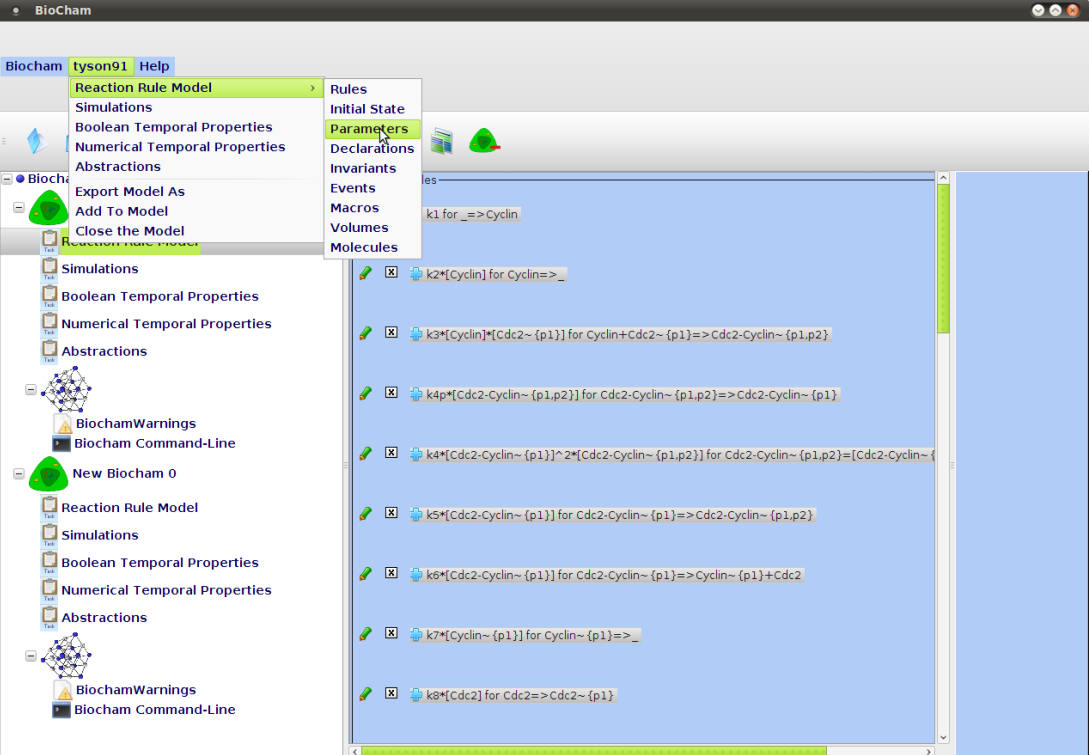
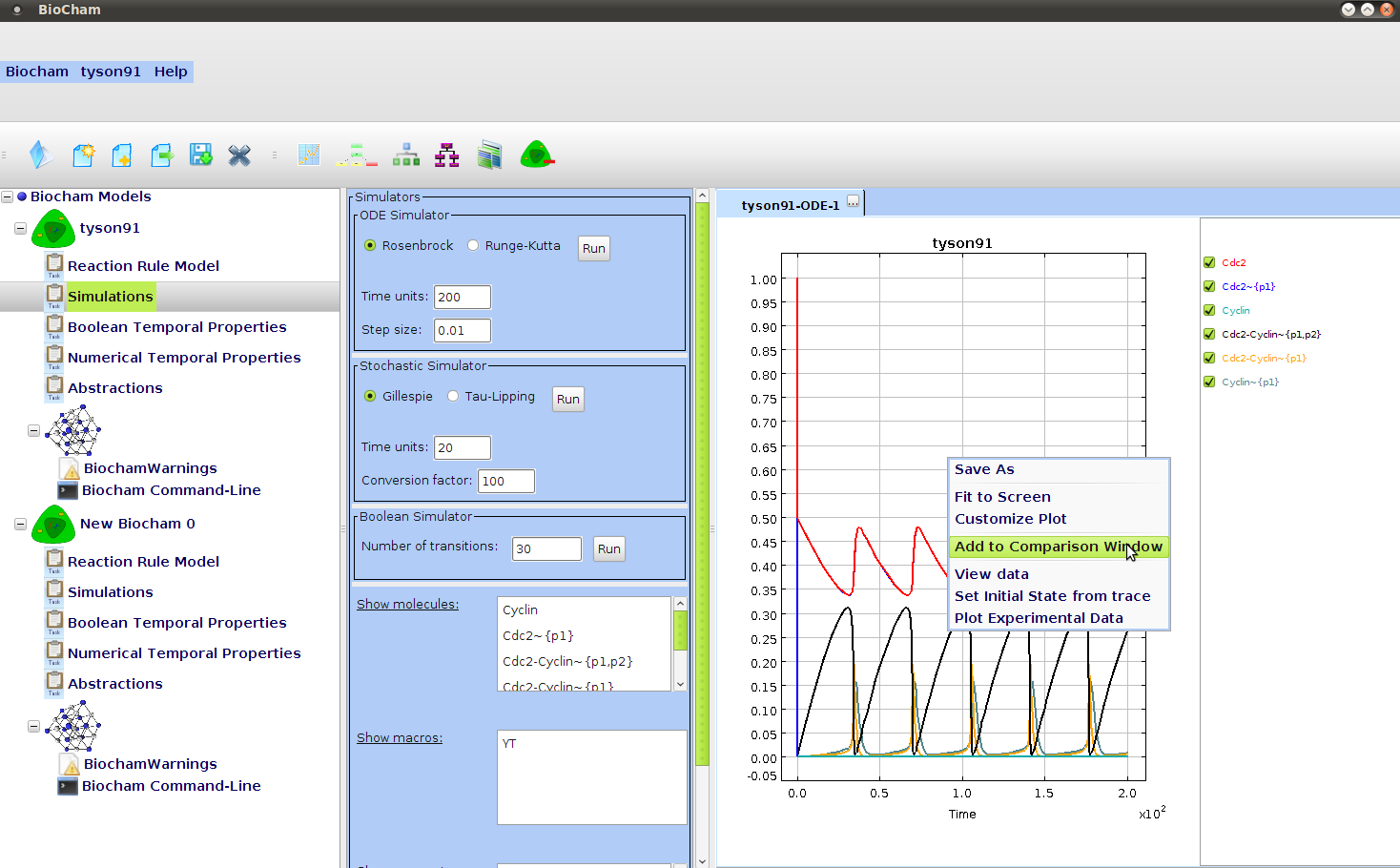
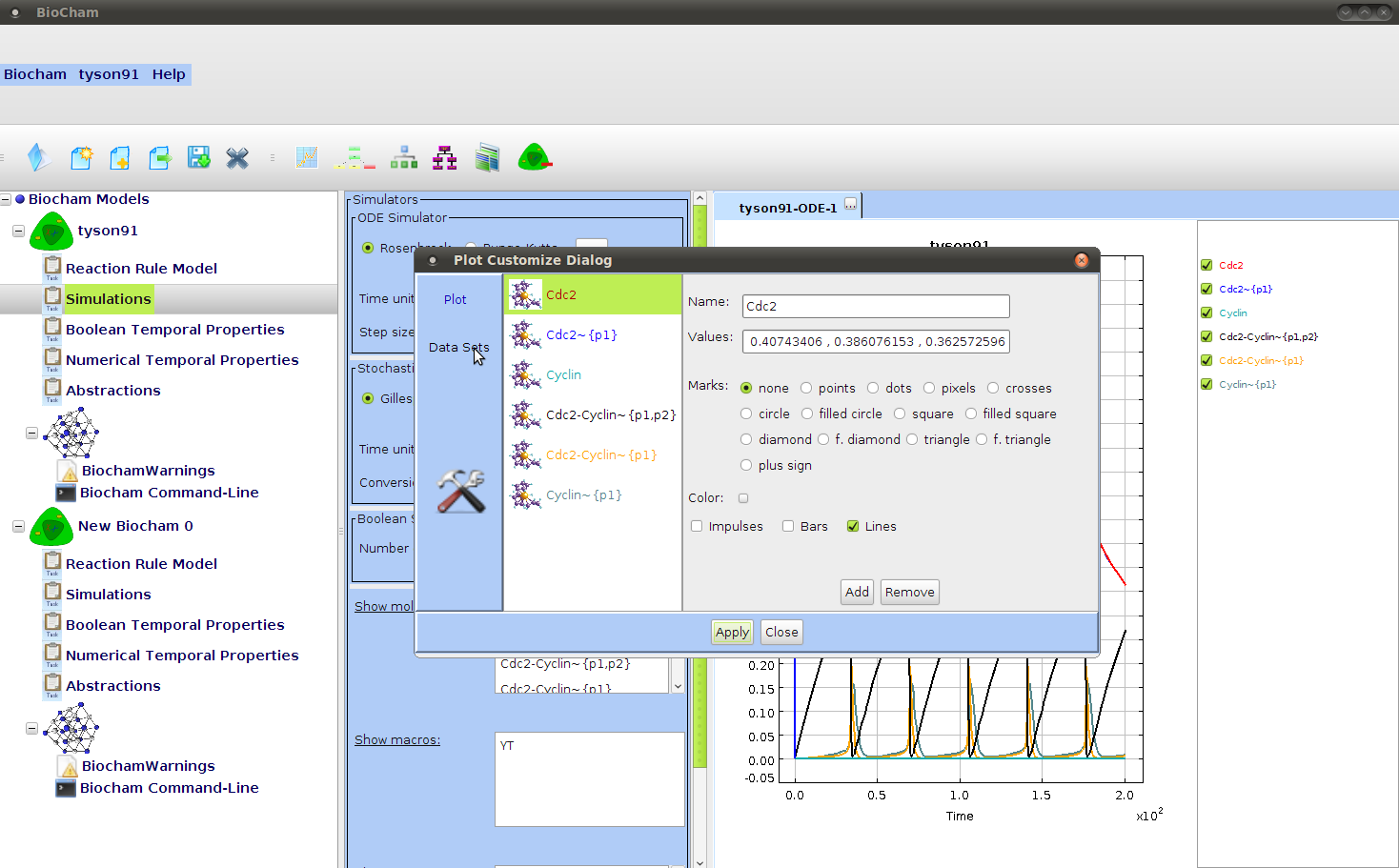
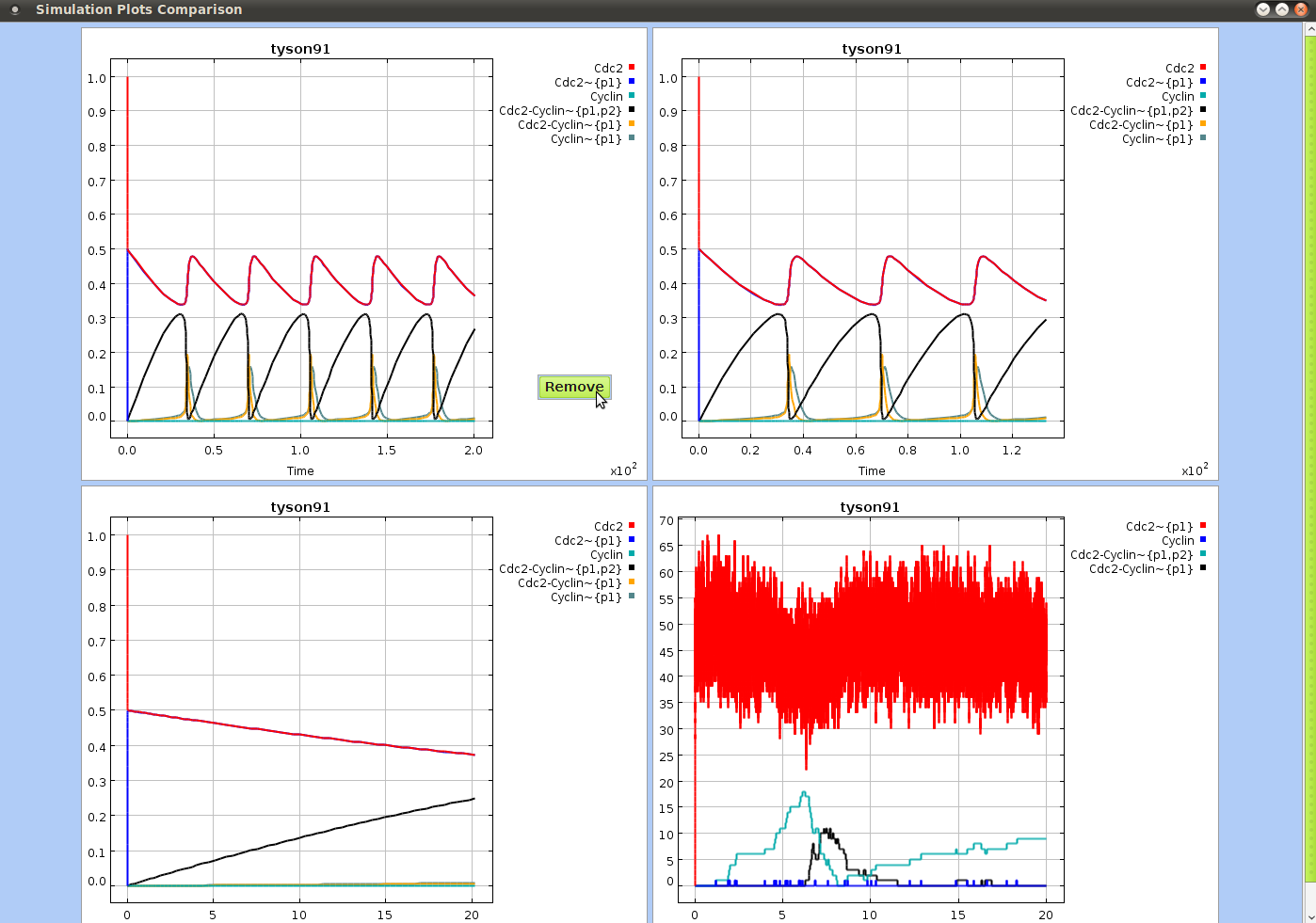
- ::, 2.2
- #, 2.2
- A, 3.1
- absent, 4.3
- add, 4.1
- add_biocham, 4.1
- add_boolean_state, 4.2
- add_boolean_transition,
4.2
- add_declaration, 4.3
- add_event, 4.2
- add_genCTL, 4.6
- add_interface, 4.2
- add_ltl, 4.7
- add_ode, 4.1
- add_rule, 4.2
- add_rules, 4.2
- add_sbml, 4.1
- add_search_condition, 4.7
- add_spec, 4.6
- add_specs, 4.6
- add_time_event, 4.2
- Ai, 3.1
- algebraic invariants, 4.4
- all, 2.5
- all_simple, 2.5
- and, 2.3, 2.5
- asynchronuous Boolean transition,
4.5
|
- biocham -debug, 1.3
- biocham -version, 1.3
- biocham_gui, 5.0
- biocham_gui -debug, 5.0
- biocham_gui -legacy, 5.4
- boolean_enumeration, 4.5
- boolean_simulation, 4.5
- catalyst, 2.3
- catalysts, 2.3
- change_directory, 4.1
- check_all_spec, 4.6
- check_conservations, 4.4
- check_ctl, 4.6
- check_ltl, 4.7
- check_ltl_spec, 4.7
- check_molecules, 4.3
- check_spec, 4.6
- check_why, 4.6
- check_why_spec, 4.6
- checkpoint, 3.1
- clear_biocham, 4.1
- clear_initial_state, 4.3
- clear_ltl, 4.7
- clear_rules, 4.2
- clear_spec, 4.6
- CMAES, 4.7
- cmaes_multi_conditions,
4.7
- cmaes_params, 4.7
- col, 1.4
- comment, 1.4
- complexation, 2.5
- compose_ode_stochastic,
4.2
- Computation Tree Logic, 3.1
- concentration, 4.5
- condition, 2.3
- conditional expression, 2.3
- conjunction, 3.1
- conservation, 4.4
- conservation laws, 4.4
- constraints, 2.5
- continue, 4.5
- continuous satisfaction degree,
4.7
- continuous-time Markov process,
4.5
- conversion_factor, 4.5
- critical_reaction_threshold,
4.5
- cross, 3.2
- csv, 1.4
- CTL, 3.1
- curate_model, 4.1
- curate_sbml, 4.1
- current_directory, 4.1
- curve_fit, 3.2
|
- debug mode, 1.3,
5.0
- declaration, 2.5
- decomplexation, 2.5
- degradation, 2.5
- delete, 4.8
- delete_conservation, 4.4
- delete_conservations, 4.4
- delete_declaration, 4.3
- delete_event, 4.2
- delete_events, 4.2
- delete_ltl, 4.7
- delete_macro, 4.4
- delete_parameter, 4.4
- delete_rules, 4.2
- delete_spec, 4.6
- delete_specs, 4.6
- delete_time_event, 4.2
- dephosphorylation, 2.5
- diff, 2.5
- dimacs, 1.4
- disjunction, 3.1
- dot, 1.4, 4.1
- draw_influences, 4.8
- draw_neighborhood, 4.8
- draw_reactions, 4.1
|
- E, 3.1
- Ei, 3.1
- elementary_interaction_rules,
2.5
- equivalence, 3.1
- event, 2.4, 4.2
- exclusive or, 3.1
- expand_biocham, 4.1
- expand_rules, 4.2
- export_biocham, 4.1
- export_biopepa, 4.1
- export_dimacs, 4.1
- export_dot, 4.1
- export_influences_dot, 4.8
- export_influences_ginml,
4.8
- export_init, 4.1
- export_lotos, 4.1
- export_neighborhood_dot,
4.8
- export_nusmv, 4.1
- export_ode, 4.1
- export_ode_latex, 4.1
- export_param, 4.1
- export_plot, 4.5
- export_prolog, 4.1
- export_sbml, 4.1
- export_sbml3, 4.1
|
- F, 3.1
- fairness_path, 4.6
- First-order LTL(R) formulae,
3.2
- first_search_condition,
4.7
- fit_x, 4.5
- fit_xmax, 4.5
- fit_xmin, 4.5
- fit_y, 4.5
- fit_ymax, 4.5
- fit_ymin, 4.5
- FOLTL(R), 3.2, 4.7
- G, 3.1
- genCTL, 4.6
- gene, 2.2
- gene knock-outs, 4.7
- gene promotor, 2.2
- get_max_from_trace, 4.5
- get_min_from_trace, 4.5
- get_period_from_trace,
4.5
- Gillespie's method, 4.5
- ginml, 1.4
- GINsim, 1.4
|
- H, 2.3
- hide_macros, 4.5
- hide_molecules, 4.5
- hide_parameters, 4.5
- Hill, 2.3
- hybrid automata, 2.4
- hybrid automaton, 2.3
- implication, 3.1
- in, 2.5
- influence graph, 4.8
- inhibitors, 2.3
- Jacobian matrix, 4.8
- json_reactions, 4.1
- keep_plot, 4.5
- kinetic expressions, 2.3
- kinetics, 2.3
- landscape, 4.7
- landscape_log, 4.7
- LaTeX, 1.4
- learn_one_addition, 4.6
- learn_one_deletion, 4.6
- Linear Time Logic, 3.2
- linking operator, 2.2
- list_all_initial_states,
4.3
- list_all_molecules, 4.3
- list_conservations, 4.4
- list_declarations, 4.3
- list_dimensions, 4.8
- list_events, 4.2
- list_functions, 4.8
- list_influences, 4.8
- list_initial_state, 4.3
- list_ltl, 4.7
- list_macros, 4.4
- list_model, 4.1
- list_molecules, 4.3
- list_neighborhood, 4.8
- list_ODE, 4.2
- list_parameters, 4.4
- list_rules, 4.2
- list_spec, 4.6
- list_volumes, 4.3
- lists, 4.0
- load, 4.1
- load_biocham, 4.1
- load_ode, 4.1
- load_sbml, 4.1
- load_trace, 4.7
- located molecule, 2.2
- location, 2.2
- loop, 3.1
- lot, 1.4
- LTL(R), 3.2, 4.7
|
- MA, 2.3
- macro, 2.2, 4.4
- make_absent_not_present,
4.3
- make_present_not_absent,
4.3
- Mass Action law, 2.3
- mass conservation law, 4.4
- merge, 4.8
- Michaelis-Menten, 2.3
- MM, 2.3
- model reductions, 4.8
- modified molecule, 2.2
- modified sites, 2.2
- molecular complex, 2.2
- molecule, 2.2
- molecule_name, 2.2
- more_elementary_interaction_rules,
2.5
- multi-trace, 4.7
- name, 2.2
- negation, 3.1
- no_fairness_path, 4.6
- no_step_doubling, 4.5
- not in, 2.5
- number of molecules, 4.5
- numerical_method, 4.5
- numerical_simulation, 4.5
- nusmv_direct, 4.6
- nusmv_disable_dynamic_reordering,
4.6
- nusmv_dynamic_reordering,
4.6
- nusmv_non_direct, 4.6
|
- object, 2.2
- object_pattern, 2.5
- ode, 1.4
- ODE interpretation, 4.5
- ordinary differential equation,
4.5
- oscil, 2.4, 3.1,
3.2
- P-invariants, 4.4
- parameter, 2.2, 4.4
- parameter_list, 4.4
- parts_of, 2.5
- pathway, 4.2
- period, 3.2
- Petri-net, 4.4
- phase_shift, 3.2
- phos_form, 2.5
- phosphorylated sites, 2.2
- phosphorylation, 2.5
- pl, 1.4
- plot, 1.4, 4.5
- png, 1.2
- positive and negative influences,
4.8
- present, 4.3
- prolog, 4.0
|
- quit, 4.0
- random, 2.3
- rdelete, 4.8
- re_complexation, 2.5
- re_phosphorylation, 2.5
- reachable, 3.1
- reaction, 2.3
- reaction_name, 2.2
- reaction_pattern, 2.5
- reduce_model, 4.6
- regulation graph, 4.8
- revise_model, 4.6
- revise_model_interactive,
4.6
- rk, 4.5
- rm_simplification_trace,
4.7
- rmerge, 4.8
- robustness, 4.7
- robustness_log, 4.7
- robustness_lognormal, 4.7
- robustness_normal, 4.7
- Rosenbrock's method, 4.5
- rule, 4.2
- Runge-Kutta method, 4.5
|
- satisfaction_degree, 4.7
- SBML, 1.4
- sdelete, 4.8
- search_all_mreductions,
4.8
- search_all_parameters, 4.7
- search_all_reductions, 4.8
- search_conservations, 4.4
- search_mreduction, 4.8
- search_parameters, 4.7
- search_parameters_cmaes,
4.7
- search_parameters_log_cmaes,
4.7
- search_random_all_parameters,
4.7
- search_random_parameters,
4.7
- search_reduction, 4.8
- seed, 2.3, 4.0,
4.7
- sensitivity, 4.7
- sensitivity_log, 4.7
- set_color, 4.5
- set_dimension, 4.8
- set_init_from_trace, 4.5
- set_initial_boolean_state,
4.2
- set_initial_state, 4.3
- set_simplification_trace,
4.7
- set_xmax, 4.5
- set_xmin, 4.5
- set_ymax, 4.5
- set_ymin, 4.5
- sets, 4.0
- show_hide, 4.5
- show_macros, 4.5
- show_molecules, 4.5
- show_parameters, 4.5
- smerge, 4.8
- smv, 1.4
- solution, 2.3
- solution_pattern, 2.5
- sq_wave, 4.4
- ssa, 4.5
- stable, 3.1
- steady, 3.1
- step_doubling, 4.5
- step_size, 4.5
- stiff, 4.5
- stochastic simulations, 4.5
- subgraph morphisms, 4.8
- submol, 2.5
- synthesis, 2.5
|
- tau-leaping, 4.5
- tau-leaping method, 4.5
- temporal Boolean properties,
3.1
- temporal quantitative properties,
3.2
- test_plot, 4.5
- Time, 2.3
- time event, 2.4
- tl, 4.5
- transition, 2.2
- true, 2.3
- U, 3.1
- undefined, 4.3
- validity domains, 4.7
- validity_domain, 4.7
- variable, 2.5
- version, 1.3
- volume, 4.3
- why, 4.6
|
- X, 3.1
- xml, 1.4
- xppaut, 1.4
|